|
MotifScan was made in Fall 2001 as a program to aid in the discovery
and visualization of protein motifs. Written entirely in Java SWING,
the program takes inputs from
TIGR genomes and MEME
results. Using MEME as the motif discovery tool from fragments isolated
from protein binding experiements and the genome and the gene coordinates
files from TIGR the program shows likely matches to the motif in all
parts of the genome and its adjacent genes.
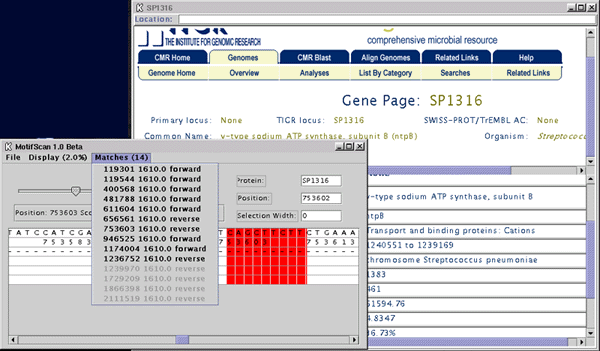
This screenshot shows the main viewing window along with the mini-browser
that aids in the exploration of genes adjacents to the selected motif.
USING
-Takes 3 Input Files
- MEME log matrix output. Generally this is from running a MEME motif
search and using the PSSM# from the motif (#) of your choice. You must
take the matrix and cut-out the first line (the one with the text) and
save it for input into MotifScan. Example:
-58 -1173 45 79
-106 209 -271 -1173
20 -1173 -1173 108
-1173 -271 145 52
-338 168 87 -1173
126 -72 -171 -180
-6 -72 119 -106
-507 162 101 -507
89 -507 -507 46
52 -271 61 -38
-1173 153 -113 20
-1173 -271 -1173 166
-1173 -1173 -1173 170
-1173 204 -39 -238
70 -72 -1173 32
This is a 15 bp motif with first column is Log-probablility of Column
1 = P(A) Column(2) = P(C) Column(3) = P(G) Column(4) = P(T).
You can also make your own Matrix for use of a concrete (you know the
sequence your looking for) or semi-concrete (pyrimidines or purines
or general idea). To do this see appendix 1.
-Sequence File. This is the whole genome that you want to search. MotifScan
assumes that the genome entered is from start to finish (e.g. from Base
1 to Base (sequence length)).
-Coords File. This file holds the start and finish coordinates for the
coding regions in the genome specified in the Sequence File. It has
the form <<Start position>> <<End position>>
<<Name>> <<junk>>. This is what .coord files
look like from TIGR.
Features:
-Contig locations. The sequence window shows the forward and reverse
coding regions (proteins). The forward strand proteins are on the row
above reverse strand proteins. The beginning of a protein is signified
with a "|" and the end is with a ">" for forward
strand and a "<" for reverse strand proteins. By clicking
on a region once, the name of that protein is displayed in the Protein
text window.
-Dragging to find distance: Click on the sequence area and hold the
button down and drag the mouse either to the right or the left. When
you let go of the mouse, the distance covered since you clicked is given
in the Selection Width Field. This facilitates finding distances to
neighboring proteins.
-Web Interface: Double Click on the sequence area on any Gene (contig)
and a mini-browser will come up referencing that gene on the TIGR website.
(This will be non-functional when genomes that are not in TIGR are used
unless the Coords file still references genes on TIGR). It works by
adding the Contig name to the link: http://www.tigr.org/tigr-scripts/CMR2/GenePage.spl?locus=
. This cannot be expected to work if the protein name attached to the
end of the link is non-existent.
-Selection Tool: Use the slider to cut-off lower scoring matches to
facilitate viewing more important matches.
-Width Tool: Enter in a width to the left of the Width button and click
on the button. From then on, the viewer will show that many bases on
both sides of the matching motif.
-Show Percent: On the menu bar, use the percent menu to show relative
amounts of matches. You can tune the amount of data you see. Use a smaller
percent to show less matches, and a larger one to show more.
Appendix 1:
To make a file that expresses a concrete motif:
Rules:
-Each line is one base pair in the motif.
-Each Column is the Log probability of getting that base at that position
-Column 1 = P(A) Column(2) = P(C) Column(3) = P(G) Column(4) = P(T).
Example:
-Since this uses log-probability, the number should be whole numbers
with their relative intensity singnifying its certainty.
Sequence: ATC
Matrix: 100 0 0 0
0 0 0 100
0 0 100 0
Sequence: A[Pyrmimidines C/T]C
Matrix: 100 0 00
0 50 0 50
0 0 100 0
Using self made matrices it is possible to formformulate your own motifs
and bypass MEME
Download the installer. Please view the
MotifScan1.doc file for help on using the program.
View Source Files
Try the Web Installer.
|